En esta entrada seleccionaremos y analizaremos una secuencia nucleotídica, teniendo en cuenta los
parámetros y herramientas que hemos estudiado a lo largo de toda la asignatura
práctica de Biosíntesis de Macromoléculas. La secuencia problema así como las cuestiones que nos proponemos determinar se encuentran accesibles en el siguiente enlace.
En primer lugar, utilizaremos el programa Chromas
Lite® que nos proporciona un cromatograma
de la secuencia problema, del que se adjunta un fragmento a continuación:

Posteriormente, exportamos la secuencia en
formato FASTA. De esta forma,
podremos trabajar con la secuencia en cualquiera de las herramientas
bioinformáticas para obtener información de las macromoléculas, en general, y
los ácidos nucleicos, en particular.
1. Análisis de secuencias con EMBOSS:
A continuación vamos a adentrarnos en la información que podemos obtener de la secuencia problema haciendo uso de JEMBOSS, una interfaz gráfica del paquete de programas EMBOSS (European Molecular Biology Open Software Suite).
Valoración de CDS con Plotorf
En primer lugar, accedemos al paquete informático JEMBOSS, que está disponible online.
Buscamos en el menú de la izquierda la opción Plotorf, que nos permitirá identificar los CDS de nuestra secuencia problema. Los resultados obtenidos se muestran a continuación:

Se nos proporcionan las seis posibles fases de lectura para la secuencia nucleotídica y, resaltado en color, los fragmentos de dicha secuencia que codifican para una proteína, de donde podemos deducir que, con mayor probabilidad, la fase de lectura para la secuencia problema es la número cuatro.
Obtención de marcos abiertos de lectura con Sixpack
A continuación, nos disponemos a traducir la secuencia a sus seis posibles fases de lectura. Para ello, haremos uso de una herramienta también de EMBOSS: EMBOSS Sixpack. Encontraréis el correspondiente enlace para acceder a ella en el menú de la izquierda de la página principal de JEMBOSS online.
Tras lanzar el Sixpack nos aparecerá una interfaz que esquematiza las secuencias peptídicas (en código de una letra) de las seis posibles fases de lectura de nuestra secuencia nucleotídica (en la siguiente imagen se muestra un fragmento de la misma). Se adjunta además, un recuento del total de ORF's (marcos de lectura abiertos) para cada una de dichas fases.


Puntuación de posibles regiones codificantes: Tcode
Para finalizar con EMBOSS vamos a emplear la herramienta Tcode. Haciendo uso de una serie de algoritmos, Tcode es capaz de identificar regiones codificantes de proteínas en una secuencia dada. Para acceder a esta herramienta basta con seguir el procedimiento llevado a cabo para el resto de programas de EMBOSS.
Tcode hace una lista de los posibles CDS de nuestra secuencia problema (que muestra finalmente en un gráfico), dándoles una puntuación (Testcode value) que le permite clasificarlos en coding (codificante, por encima de la línea verde), non- coding (no codificante, por debajo de la línea roja) o no opinion (puede tratarse de un fragmento codificante o no codificante, entre las líneas verde y roja).

2. Comparación de secuencias:
Anotaciones:
Como hemos podido comprobar en entradas anteriores, una de las herramientas más útiles para la comparación de secuencias es BLAST (Basic Local Alignment Search Tool), un programa bioinformático de alineamiento de secuencias de tipo local (ADN, ARN, proteínas).
Podemos distinguir entre varias modalidades de BLAST. En este caso realizaremos BlastN (Nucleotide Blast), que compara una secuencia nucleotídica con bases de datos que contengan también secuencias nucleotídicas.
Se nos muestra, en un diagrama de barras con código de colores (representativo de la puntuación de alineamiento), las secuencias más similares como una distribución de alineamientos: cada barra representa una secuencia, ordenadas por porcentaje decreciente de similitud.
2. Comparación de secuencias:
Anotaciones:
Anotaciones:
Como hemos podido comprobar en entradas anteriores, una de las herramientas más útiles para la comparación de secuencias es BLAST (Basic Local Alignment Search Tool), un programa bioinformático de alineamiento de secuencias de tipo local (ADN, ARN, proteínas).
Podemos distinguir entre varias modalidades de BLAST. En este caso realizaremos BlastN (Nucleotide Blast), que compara una secuencia nucleotídica con bases de datos que contengan también secuencias nucleotídicas.
Se nos muestra, en un diagrama de barras con código de colores (representativo de la puntuación de alineamiento), las secuencias más similares como una distribución de alineamientos: cada barra representa una secuencia, ordenadas por porcentaje decreciente de similitud.

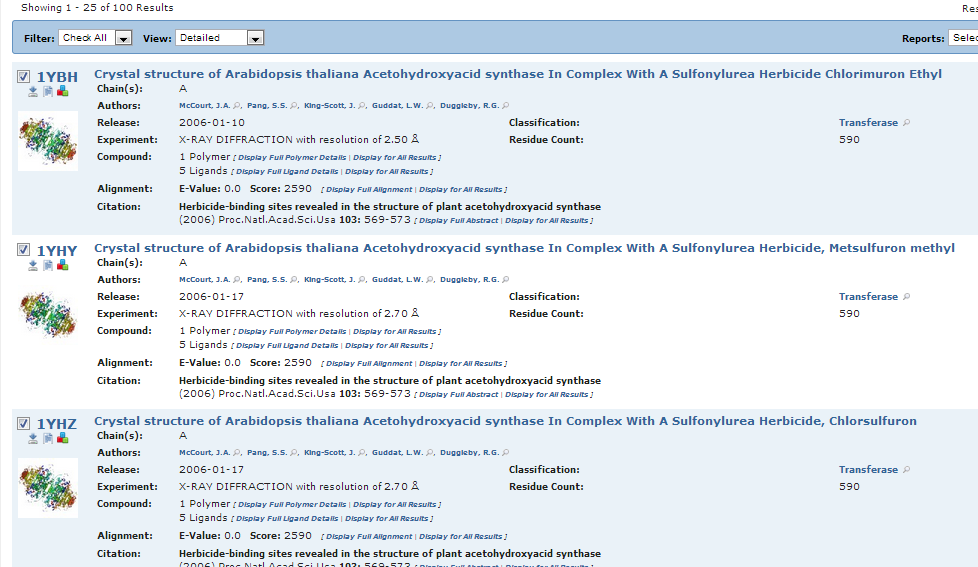
La primera de las barras (máxima similitud) se corresponde con el CDS completo de la proteína ROS1 de Arabidopsis thaliana, por lo que deducimos que nuestra secuencia problema se corresponde con el gen que codifica dicha proteína.
La primera de las barras (máxima similitud) se corresponde con el CDS completo de la proteína ROS1 de Arabidopsis thaliana, por lo que deducimos que nuestra secuencia problema se corresponde con el gen que codifica dicha proteína.
3. Información sobre la proteína:
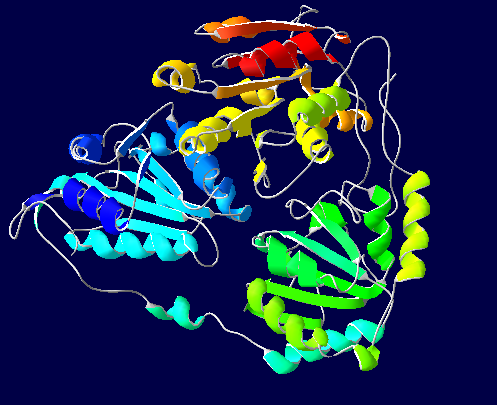
Clickamos en el número de accesión correspondiente a dicha proteína (NM_129207.4) y accedemos a una interfaz que nos proporciona mayor información (locus del gen, clasificación del organismo, artículo y publicación...), además se muestra la secuencia proteínica (en código de una letra) deducible a partir de nuestra secuencia nucleotídica problema, y que se adjunta a continuación:

3. Información sobre la proteína:
Clickamos en el número de accesión correspondiente a dicha proteína (NM_129207.4) y accedemos a una interfaz que nos proporciona mayor información (locus del gen, clasificación del organismo, artículo y publicación...), además se muestra la secuencia proteínica (en código de una letra) deducible a partir de nuestra secuencia nucleotídica problema, y que se adjunta a continuación:
Clickamos en el número de accesión correspondiente a dicha proteína (NM_129207.4) y accedemos a una interfaz que nos proporciona mayor información (locus del gen, clasificación del organismo, artículo y publicación...), además se muestra la secuencia proteínica (en código de una letra) deducible a partir de nuestra secuencia nucleotídica problema, y que se adjunta a continuación:
Entre los aspectos más relevantes que podemos encontrar en las anotaciones, se encuentra la información relativa a la funcionalidad de la proteína. En este caso en particular, encontramos que nuestra proteína se trata de un represor transcripcional que actúa en procesos de silenciameiento génico, mediante la desmetilación de regiones del promotor diana. Interacciona físicamente con otras proteínas, como RPA2/ROR1. Se ha encontrado en mutantes de Ros1 un aumento de la metilación en varios promotores.
Entre los loci afectados por ros1, algunos se ven afectados en la metilación de citosinas aunque la mayor parte de ellos se afectan en otros nucleótidos.
En la interfaz de las anotaciones aparece un menú a la derecha con enlaces a varias bases de datos donde podremos completar la búsqueda y ampliar la información. Una de las opciones es Map viewer, que de una forma gráfica sitúa nuestro gen en su correspondiente cromosoma (en este caso, cromosoma 2), junto con el resto de loci de dicho cromosoma, pudiendo saber qué genes se encuentran más cerca de ros1 y, por tanto, con una alta probabilidad segregarán juntos.
InterPro:
A continuación, vamos a hacer uso de Interpro (en concreto, Interproscan) para identificar dominios de interés en la proteína ROS1.
Para ello accedemos en primer lugar a la página web de Interpro, haciendo click en el enlace, y buscamos la herramienta InterproScan y copiamos la secuencia peptídica que obtuvimos al realizar el Blast en el recuadro de inserción, clickamos en submit y ya sólo queda esperar los resultados.

Encontramos esquematizadas las distintas regiones con dominios, mostradas bajo un código de colores (según la base de datos de procedencia). Observamos algunos dominios HHH (varias hélices alfa) y HTH (hélice-giro-hélice) que se relacionan con una DNA glicosilasa (acción de reparación del daño en el ADN) que podemos inferir que pertenece a la familia de endonucleasas III.
Haciendo click en los recuadros al comienzo de cada representación, obtendremos información más detallada, así como los números de accesión GO.
BlastX y BlastP
Para finalizar esta práctica resumen vamos a realizar dos útlimos Blast:
- Blastx: este programa usa como entrada una secuencia nucleotídica que traduce en sus seis posibles marcos de lectura, y compara estas secuencias con proteínas recogidas en bases de de datos (suele utilizarse cuando se sospecha que la secuencia de entrada codifica para una proteína, pero no se sabe exactamente cuál es su producto).
Los resultados que obtenemos tras lanzar el Blastx se esquematizan a continuación:
 Obtenemos que la secuencia con mayor porcentaje de similitud se corresponde con la proteína Ros1 de Arabidpsis thaliana, como era de esperar. El resto de líneas que aparacen en el Blast hacen referencia a proteínas hipotéticas con secuencia recogida en las bases de datos.
Obtenemos que la secuencia con mayor porcentaje de similitud se corresponde con la proteína Ros1 de Arabidpsis thaliana, como era de esperar. El resto de líneas que aparacen en el Blast hacen referencia a proteínas hipotéticas con secuencia recogida en las bases de datos.
- BlastP: este programa compara una secuencia de aminoácidos con otras secuencias, también de aminoácidos, recogidas en bases de datos, para realizar alineamientos introduciendo gaps (huecos) según matrices de sustitución BLOSUM o PAM.
Tras realizar el BlastP obtenemos los siguientes resultados:
 En este caso, obtenemos un mayor número de coincidencias. Las dos primeras secuencias se corresponden con secuencias de la proteína ROS1 de Arabidopsis, almacenadas en distintas bases de datos. Del mismo modo que cuando llevamos a cabo el BlastX, el resto de coincidencias (con menor porcentaje de similitud) se corresponde con secuencias catalogadas como proteínas hipotéticas.
En este caso, obtenemos un mayor número de coincidencias. Las dos primeras secuencias se corresponden con secuencias de la proteína ROS1 de Arabidopsis, almacenadas en distintas bases de datos. Del mismo modo que cuando llevamos a cabo el BlastX, el resto de coincidencias (con menor porcentaje de similitud) se corresponde con secuencias catalogadas como proteínas hipotéticas.
Entre los aspectos más relevantes que podemos encontrar en las anotaciones, se encuentra la información relativa a la funcionalidad de la proteína. En este caso en particular, encontramos que nuestra proteína se trata de un represor transcripcional que actúa en procesos de silenciameiento génico, mediante la desmetilación de regiones del promotor diana. Interacciona físicamente con otras proteínas, como RPA2/ROR1. Se ha encontrado en mutantes de Ros1 un aumento de la metilación en varios promotores.
Entre los loci afectados por ros1, algunos se ven afectados en la metilación de citosinas aunque la mayor parte de ellos se afectan en otros nucleótidos.
En la interfaz de las anotaciones aparece un menú a la derecha con enlaces a varias bases de datos donde podremos completar la búsqueda y ampliar la información. Una de las opciones es Map viewer, que de una forma gráfica sitúa nuestro gen en su correspondiente cromosoma (en este caso, cromosoma 2), junto con el resto de loci de dicho cromosoma, pudiendo saber qué genes se encuentran más cerca de ros1 y, por tanto, con una alta probabilidad segregarán juntos.
InterPro:
A continuación, vamos a hacer uso de Interpro (en concreto, Interproscan) para identificar dominios de interés en la proteína ROS1.
Para ello accedemos en primer lugar a la página web de Interpro, haciendo click en el enlace, y buscamos la herramienta InterproScan y copiamos la secuencia peptídica que obtuvimos al realizar el Blast en el recuadro de inserción, clickamos en submit y ya sólo queda esperar los resultados.
Encontramos esquematizadas las distintas regiones con dominios, mostradas bajo un código de colores (según la base de datos de procedencia). Observamos algunos dominios HHH (varias hélices alfa) y HTH (hélice-giro-hélice) que se relacionan con una DNA glicosilasa (acción de reparación del daño en el ADN) que podemos inferir que pertenece a la familia de endonucleasas III.
Haciendo click en los recuadros al comienzo de cada representación, obtendremos información más detallada, así como los números de accesión GO.
BlastX y BlastP
Para finalizar esta práctica resumen vamos a realizar dos útlimos Blast:
- Blastx: este programa usa como entrada una secuencia nucleotídica que traduce en sus seis posibles marcos de lectura, y compara estas secuencias con proteínas recogidas en bases de de datos (suele utilizarse cuando se sospecha que la secuencia de entrada codifica para una proteína, pero no se sabe exactamente cuál es su producto).
Los resultados que obtenemos tras lanzar el Blastx se esquematizan a continuación:
Obtenemos que la secuencia con mayor porcentaje de similitud se corresponde con la proteína Ros1 de Arabidpsis thaliana, como era de esperar. El resto de líneas que aparacen en el Blast hacen referencia a proteínas hipotéticas con secuencia recogida en las bases de datos.
- BlastP: este programa compara una secuencia de aminoácidos con otras secuencias, también de aminoácidos, recogidas en bases de datos, para realizar alineamientos introduciendo gaps (huecos) según matrices de sustitución BLOSUM o PAM.
Tras realizar el BlastP obtenemos los siguientes resultados:
En este caso, obtenemos un mayor número de coincidencias. Las dos primeras secuencias se corresponden con secuencias de la proteína ROS1 de Arabidopsis, almacenadas en distintas bases de datos. Del mismo modo que cuando llevamos a cabo el BlastX, el resto de coincidencias (con menor porcentaje de similitud) se corresponde con secuencias catalogadas como proteínas hipotéticas.